第一原理計算は半導体材料のバンドギャップ、欠陥形成エネルギーなどを予測できるツールです。材料開発におけるスクリーニングを計算機上で行うことで開発期間の短縮が可能になります。キャリアのナノ的な挙動を理解するためには理論計算が不可欠です。
半導体材料開発における計算科学の必要性
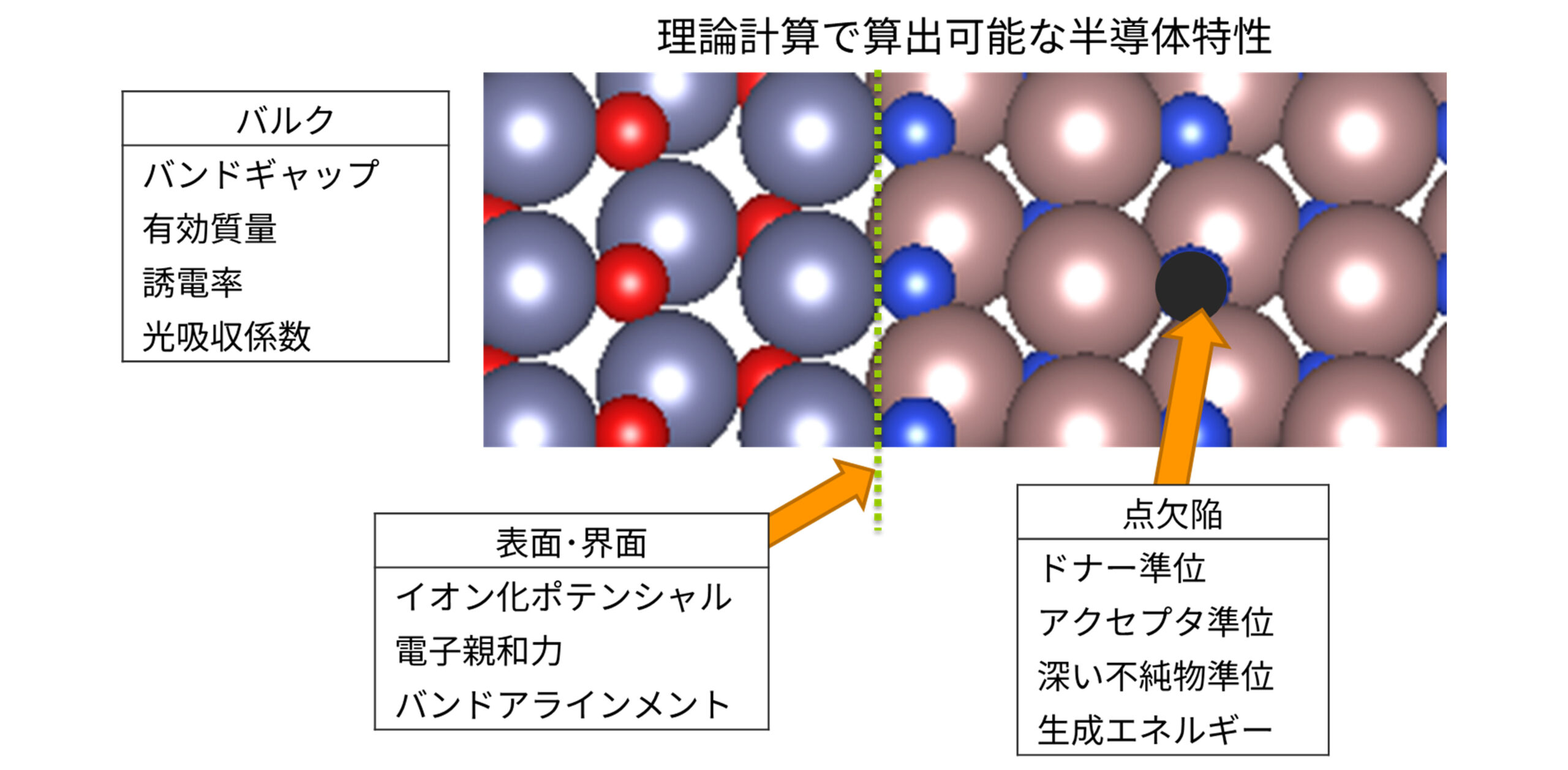
計算科学、特に第一原理計算は半導体材料のバルク、点欠陥、表面・界面にわたる物理的特性を任意の組成や構造について予測できる強力なツールです。材料開発初期におけるスクリーニングを計算機上で仮想的に行うことで、開発期間の大幅な短縮が可能になります。
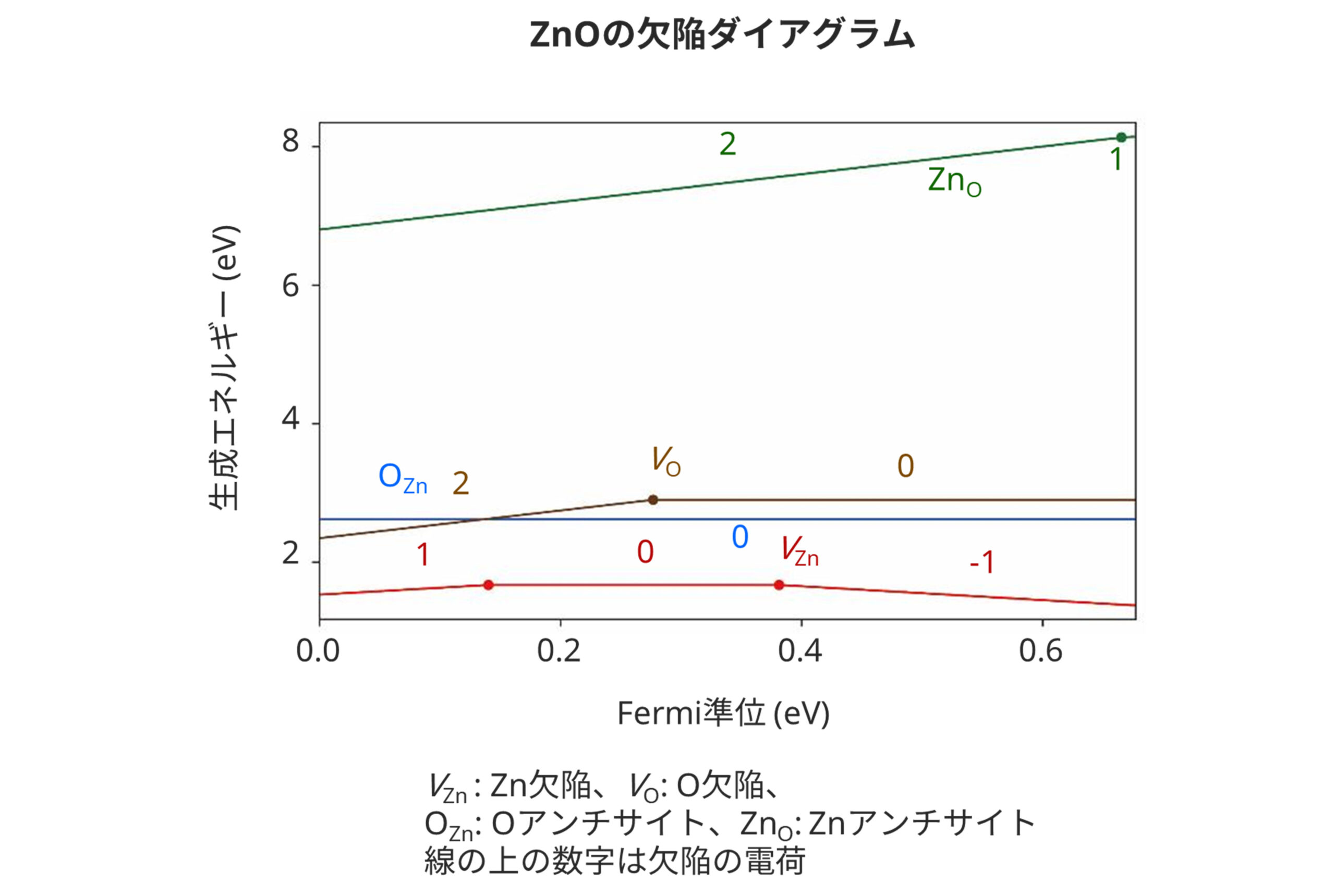
第一原理計算による欠陥ダイアグラムの作成
点欠陥の形成エネルギーを求め、Fermi準位の変化による荷電欠陥のエネルギー変化までを考慮すると「欠陥ダイアグラム」を描くことができます。この図からは欠陥濃度や、欠陥が電子を捕獲・放出するFermiエネルギー、平衡Fermi準位など、点欠陥に関わる特性を推定することができます。
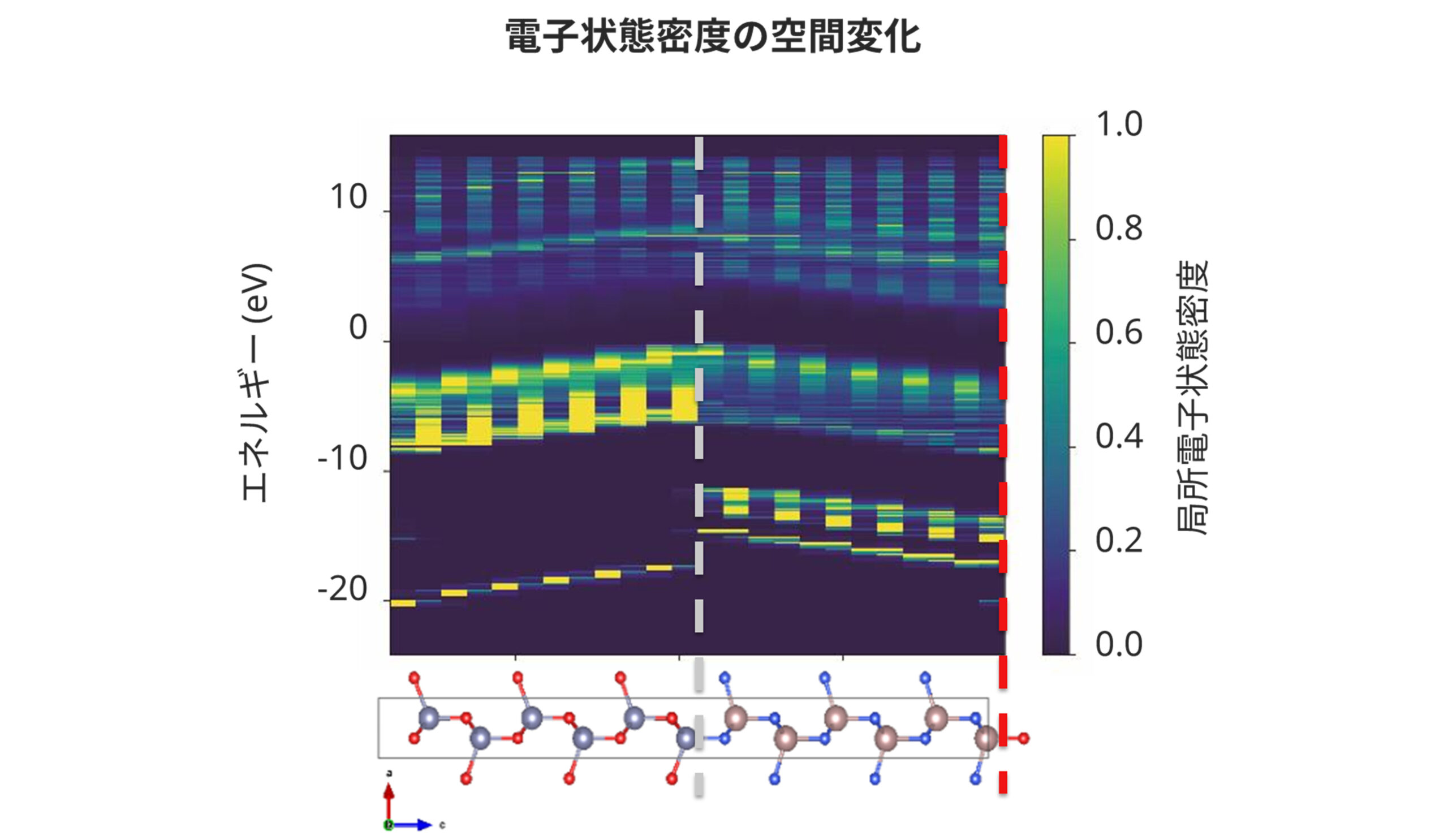
第一原理計算によるバンドベンディングの可視化
第一原理計算では各原子ごとに分解された電子状態密度を求めることができ、空間座標とエネルギー準位を対応づけることで接合界面のバンドベンディングを可視化することができます。下の図はZnO (左側) とGaN (右側) の界面について図示したものであり、周期境界条件下での二つの界面 (Zn-N結合:灰色破線、Ga-O結合:赤色破線) の存在によってバンドが湾曲する様子が見えています。
掲載資料をダウンロードできます。
PDF形式
左のアイコンをクリックすると、別ウインドウで開きます。
資料のダウンロードにはお客様情報の入力が必要となります。